As practice for using gradient boosted trees, I used an open dataset to use gene expression in tumours to predict resistance to chemotherapy drugs
Published
2024-04-02
Show the code
import pandas as pdimport numpy as npimport matplotlib.pyplot as pltimport lightgbmimport shapfrom sklearn.model_selection import train_test_splitfrom sklearn.metrics import mean_absolute_errorfrom sklearn.linear_model import LinearRegressionfrom re import sub
This is a bit of work I did during my postdoc at the University of Nottingham. There’s not enough here for a publication, and was a bit out of the scope of the project, so I’m writing it up here. I’m not reproducing the entirety of this work here, mostly because running the whole thing takes ages. The full workflow is still in the code, I have just commented parts of it out. If you do want to run the full analysis yourself, you can uncomment these lines and change a few variable names.
I went to a conference1 and heard a talk2 in which the speaker described using a dataset of tumour cell lines to identify proteins important in cancer to identify a target to study. The Genomics of Drug Sensitivity in Cancer dataset is a massive project testing the sensitivity of tumour cell lines to different drugs. Part of the dataset is a table describing how sensitive cell lines are to each of a few hundred drugs.
Show the code
# It seems that previous releases get moved, so I'm just downloading it locally instead of getting it via ftp#ic50_data = pd.read_excel("ftp://ftp.sanger.ac.uk/pub/project/cancerrxgene/releases/current_release/GDSC1_fitted_dose_response_25Feb20.xlsx")ic50_data = pd.read_excel("data/GDSC1_fitted_dose_response_27Oct23.xlsx")ic50_data.drop(columns = ['NLME_RESULT_ID', 'NLME_CURVE_ID', 'SANGER_MODEL_ID', 'TCGA_DESC', 'DRUG_ID', 'PUTATIVE_TARGET', 'PATHWAY_NAME', 'COMPANY_ID', 'WEBRELEASE', 'MIN_CONC', 'MAX_CONC', 'AUC', 'RMSE'], inplace=True)ic50_data.head()
DATASET
COSMIC_ID
CELL_LINE_NAME
DRUG_NAME
LN_IC50
Z_SCORE
0
GDSC1
684057
ES5
Erlotinib
3.966813
1.299144
1
GDSC1
684059
ES7
Erlotinib
2.692090
0.156076
2
GDSC1
684062
EW-11
Erlotinib
2.477990
-0.035912
3
GDSC1
684072
SK-ES-1
Erlotinib
2.033564
-0.434437
4
GDSC1
687448
COLO-829
Erlotinib
2.966007
0.401702
I’m just using the first version of the dataset here, as shown by “GDSC1” in the DATASET column. There is a GDSC2 that could be added, but we have enough to be getting on with. The COSMIC_ID and CELL_LINE_NAME columns identify the cell line used in the experiment. DRUG_NAME should be fairly obvious.
The last two columns might be a bit less familiar. To explain these, I will have to be a little more precise about what “sensitivity” means. To create this dataset, the experimenters grew lots of samples of each cell line, and added different concentrations of drugs to the samples. They then let them grow for a bit, and counted how many were still viable (alive and able to keep going). They then analysed this data to see how much each concentration of the drug slowed the growth of cells. They then fitted this to a dose response curve to get an estimate of the concentration of the drug that inhibited growth by 50, a measure called the IC50. This measure is best described using a natural logarithm, so this is reported as LN_IC50. If you want to get a better feel for dose-response curves, I have a toy here. The Z_SCORE describes how significant this difference is for that cell line, compared with the sensitivity of other cell lines to that drug.
Another part of the dataset is a measure of how genes are expressed in each cell line. For each of the cell lines in the dataset, the mRNA for each gene has been measured using a microarray.
Show the code
# I did have it downloading directly from the source, but I've had failures retrieving it and the IC50 data, so I'm loading it locally instead#rma_expr = pd.read_csv("https://www.cancerrxgene.org/gdsc1000/GDSC1000_WebResources//Data/preprocessed/Cell_line_RMA_proc_basalExp.txt.zip", sep = "\t")rma_expr = pd.read_csv("data/Cell_line_RMA_proc_basalExp.txt", sep ="\t")rma_expr = rma_expr.drop('GENE_title', axis =1)rma_expr= rma_expr.set_index('GENE_SYMBOLS')rma_expr.head()
DATA.906826
DATA.687983
DATA.910927
DATA.1240138
DATA.1240139
DATA.906792
DATA.910688
DATA.1240135
DATA.1290812
DATA.907045
...
DATA.753584
DATA.907044
DATA.998184
DATA.908145
DATA.1659787
DATA.1298157
DATA.1480372
DATA.1298533
DATA.930299
DATA.905954.1
GENE_SYMBOLS
TSPAN6
7.632023
7.548671
8.712338
7.797142
7.729268
7.074533
3.285198
6.961606
5.943046
3.455951
...
7.105637
3.236503
3.038892
8.373223
6.932178
8.441628
8.422922
8.089255
3.112333
7.153127
TNMD
2.964585
2.777716
2.643508
2.817923
2.957739
2.889677
2.828203
2.874751
2.686874
3.290184
...
2.798847
2.745137
2.976406
2.852552
2.622630
2.639276
2.879890
2.521169
2.870468
2.834285
DPM1
10.379553
11.807341
9.880733
9.883471
10.418840
9.773987
10.264385
10.205931
10.299757
11.570155
...
10.486486
10.442951
10.311962
10.454830
10.418475
11.463742
10.557777
10.792750
9.873902
10.788218
SCYL3
3.614794
4.066887
3.956230
4.063701
4.341500
4.270903
5.968168
3.715033
3.848112
5.560883
...
3.696835
4.624013
4.348524
3.858121
3.947561
4.425849
3.550390
4.443337
4.266828
4.100493
C1orf112
3.380681
3.732485
3.236620
3.558414
3.840373
3.815055
3.011867
3.268449
3.352835
3.571228
...
3.726833
3.947744
3.806584
3.196988
3.814831
4.384732
4.247189
3.071359
3.230197
3.435795
5 rows × 1018 columns
In this table, each row is a gene, and each column shows the expression of that gene in a cell line. This data can be used with the drug sensitivity dataset using the COSMIC_ID column of that dataset. The column names in the expression table contain the COSMIC ID (DATA.[COSMIC ID]).
We can see how many of the cell lines described in the expression table are present in the sensitivity table.
Show the code
rma_cells = [int(x.split('.')[1]) for x in rma_expr.columns]cell_id_matches = []for cell_id in rma_cells:if cell_id in ic50_data['COSMIC_ID'].values: cell_id_matches.append(cell_id)print(f'Number of matches: {len(cell_id_matches)} of {len(rma_cells)}')
Number of matches: 946 of 1018
And filter the expression data so we’re only using columns that are relevant to the cell lines of interest.
What’s the plan here? A lot of statistical analysis has been carried out on this dataset by bioinformaticians much smarter than I am. What I want to get an idea of is how well this dataset can be used to predict drug sensitivity from expression data. In this first pass, I’m getting a baseline. There are things you can do to improve models like this, which I have carried out, but let’s pretend, for the sake of narrative, that I haven’t yet.
To use this data to train a predictive model, we really want a table where each row is a cell line, and each column is a gene.
Show the code
rma_expr_matched = rma_expr_matched.Trma_expr_matched['COSMIC_ID'] = [int(x.split('.')[1]) for x in rma_expr_matched.index]rma_expr_matched = rma_expr_matched.loc[:, rma_expr_matched.columns.notna()]rma_expr_matched.head()
GENE_SYMBOLS
TSPAN6
TNMD
DPM1
SCYL3
C1orf112
FGR
CFH
FUCA2
GCLC
NFYA
...
OR1D5
ZNF234
MYH4
LINC00526
PPY2
KRT18P55
POLRMTP1
UBL5P2
TBC1D3P5
COSMIC_ID
DATA.906826
7.632023
2.964585
10.379553
3.614794
3.380681
3.324692
3.566350
8.204530
5.235118
5.369039
...
3.134197
4.841169
2.628932
6.786925
2.997054
3.331134
3.130696
9.986616
3.073724
906826
DATA.687983
7.548671
2.777716
11.807341
4.066887
3.732485
3.152404
7.827172
6.616972
5.809264
7.209653
...
3.327528
4.570476
2.783441
5.317911
3.263745
2.992611
3.260982
9.002814
3.000182
687983
DATA.910927
8.712338
2.643508
9.880733
3.956230
3.236620
3.241246
2.931034
8.191246
5.426841
5.120747
...
3.326309
4.214729
2.603604
3.143006
3.112145
2.886574
3.176239
9.113243
2.916274
910927
DATA.1240138
7.797142
2.817923
9.883471
4.063701
3.558414
3.101247
7.211707
8.630643
5.617714
4.996434
...
2.921903
4.060761
2.619540
3.153896
3.151576
3.812119
3.074432
9.958284
3.256500
1240138
DATA.1240139
7.729268
2.957739
10.418840
4.341500
3.840373
3.001802
3.375422
8.296950
5.669418
4.180205
...
3.474086
4.869199
2.450375
3.652660
2.918475
3.412586
3.213545
9.938978
3.396126
1240139
5 rows × 17420 columns
I’m going to use a kind of model called “Gradient Boosted Trees” to predict drug sensitivity here. I won’t go into too much detail on how they work (Google has a course on them here), but essentially they build up a model from a set of small models called decision trees. It starts with a simple decision tree, then adds another simple decision tree to the output of that one that is chosen to best deal with the shortcomings of the previous. This process is repeated until adding more trees doesn’t improve prediction very much. These models perform really well on small, structured datasets, so are a good choice here. The implementation I’m using is called LightGBM, which, from my understanding, makes models that are nearly as good as gradient boosted trees can be, but are relatively fast.
For each drug, my model_drug function takes the IC50 data matching that drug, then merges that data with matching data from the expression table.
It then splits this into a training and test set. The training set contains 80% of the data. The decision trees are built based on this data, then the model’s performance is assessed on how well it predicts the IC50 in the remaining 20%.
In my first run at this, I trained a model for each of the drugs in the dataset. To do this, I had to start it when I got in one morning, go do a day’s work in the lab, then come back to collect the results. Here, I’ve just chosen 14 drugs that (spoiler alert) do well here, and one that I know doesn’t. If you want to check my work, the code for running the whole lot is still in there.
For each drug, the Mean Absolute Error of the model and the range of ln(IC50) in the test set are reported.
AZ628:
MAE = 1.23 (range 8.49)
WZ3105:
MAE = 1.19 (range 8.98)
NPK76-II-72-1:
MAE = 1.07 (range 7.88)
Tubastatin A:
MAE = 0.858 (range 6.29)
PIK-93:
MAE = 1.13 (range 8.7)
Venotoclax:
MAE = 0.629 (range 9.47)
Methotrexate:
MAE = 1.0 (range 7.75)
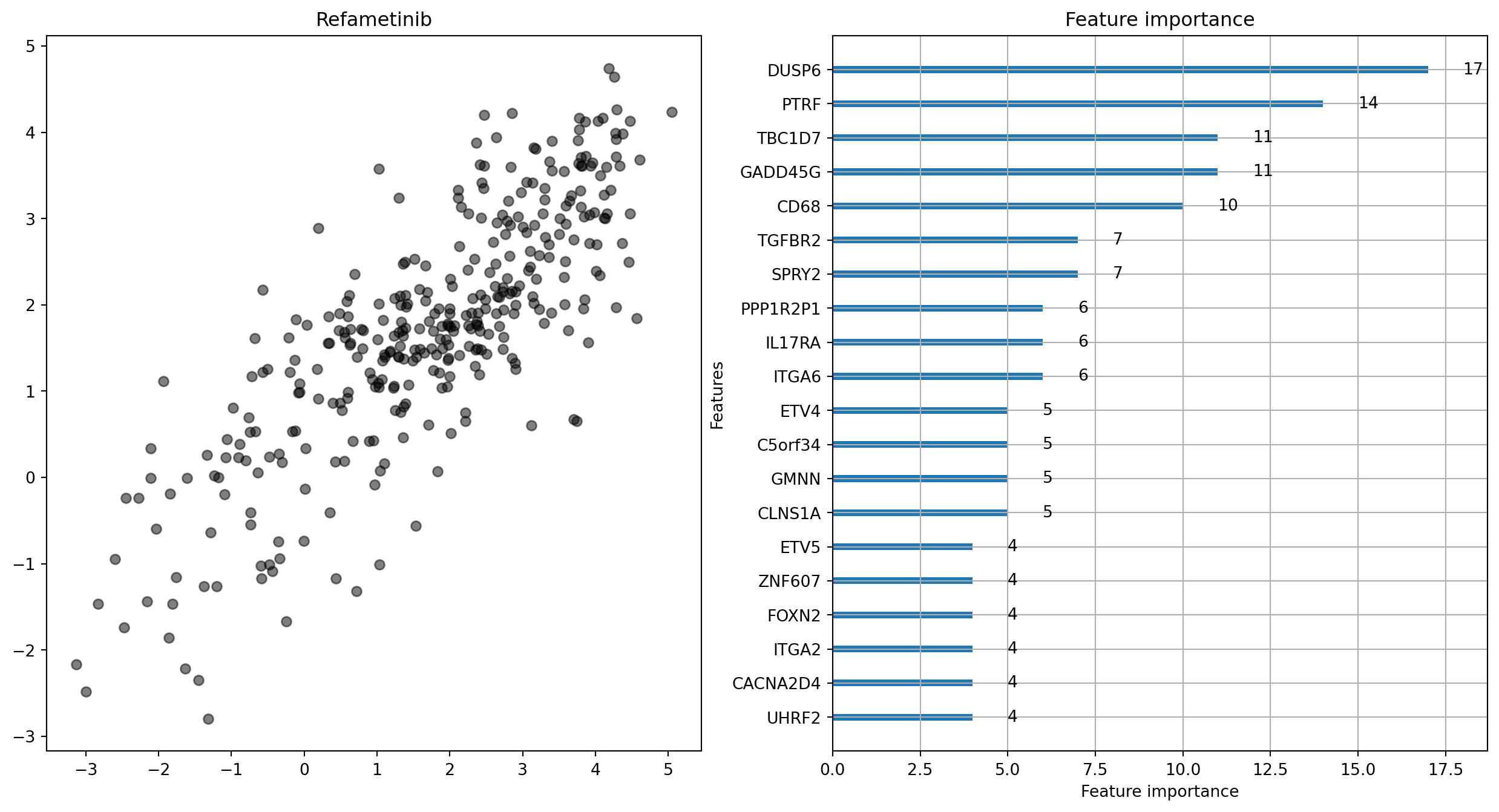
Refametinib:
MAE = 0.811 (range 8.17)
AZD7762:
MAE = 0.785 (range 10.7)
Tanespimycin:
MAE = 0.961 (range 7.35)
Nutlin-3a (-):
MAE = 0.741 (range 5.03)
Trametinib:
MAE = 1.39 (range 10.4)
Dabrafenib:
MAE = 1.26 (range 10.3)
SN-38:
MAE = 1.01 (range 10.6)
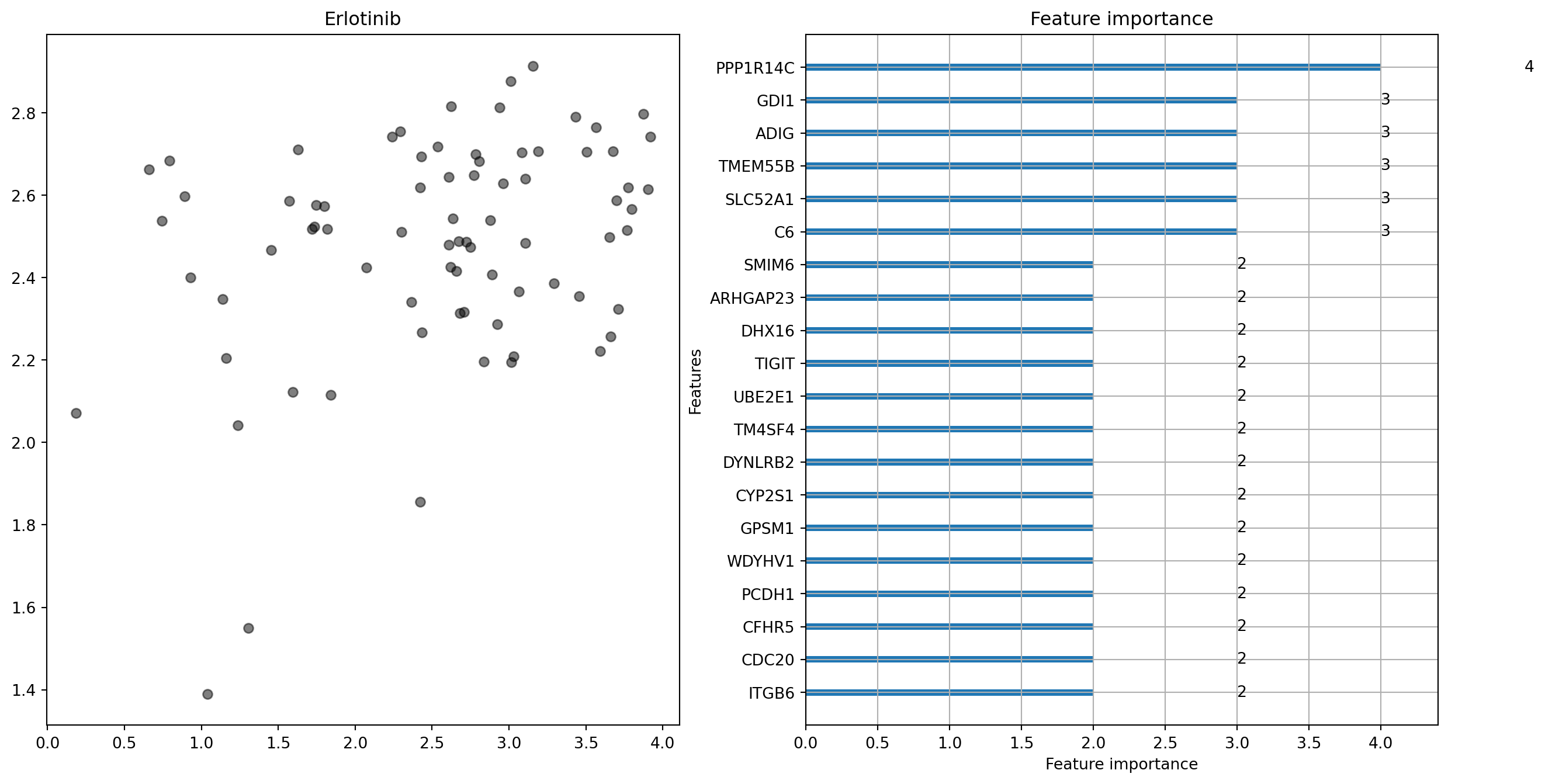
Erlotinib:
MAE = 0.699 (range 3.74)
The R2 value describes how much of the variance of a dependent variable can be explained by the independent variable. I’m using it here to get an idea of how well the predictions from the models correlate with the observations.
Show the code
def r_squared(predicted, true): mean = np.mean(true) true_diff_sq = np.square(true - mean) pred_diff_sq = np.square(true - predicted)return1-(np.sum(pred_diff_sq)/np.sum(true_diff_sq))models_r_sq =dict([(x, r_squared(y[1], y[2])) for x, y in example_models.items()])[(x,y) for x,y in models_r_sq.items() if y >0.4]
And for Erlotinib, one that can’t (in this first go, at least).
Show the code
plot_test('Erlotinib')
The panels on the left of these plots show this comparison. Hopefully, you’ll be wondering what the other panel shows. A neat feature of LightGBM models is that the model can tell you how important different dependent variables are when making a prediction. This means that we can get an idea of how the differential expression of particular genes contributes to the sensitivity of tumour cells to drugs3.
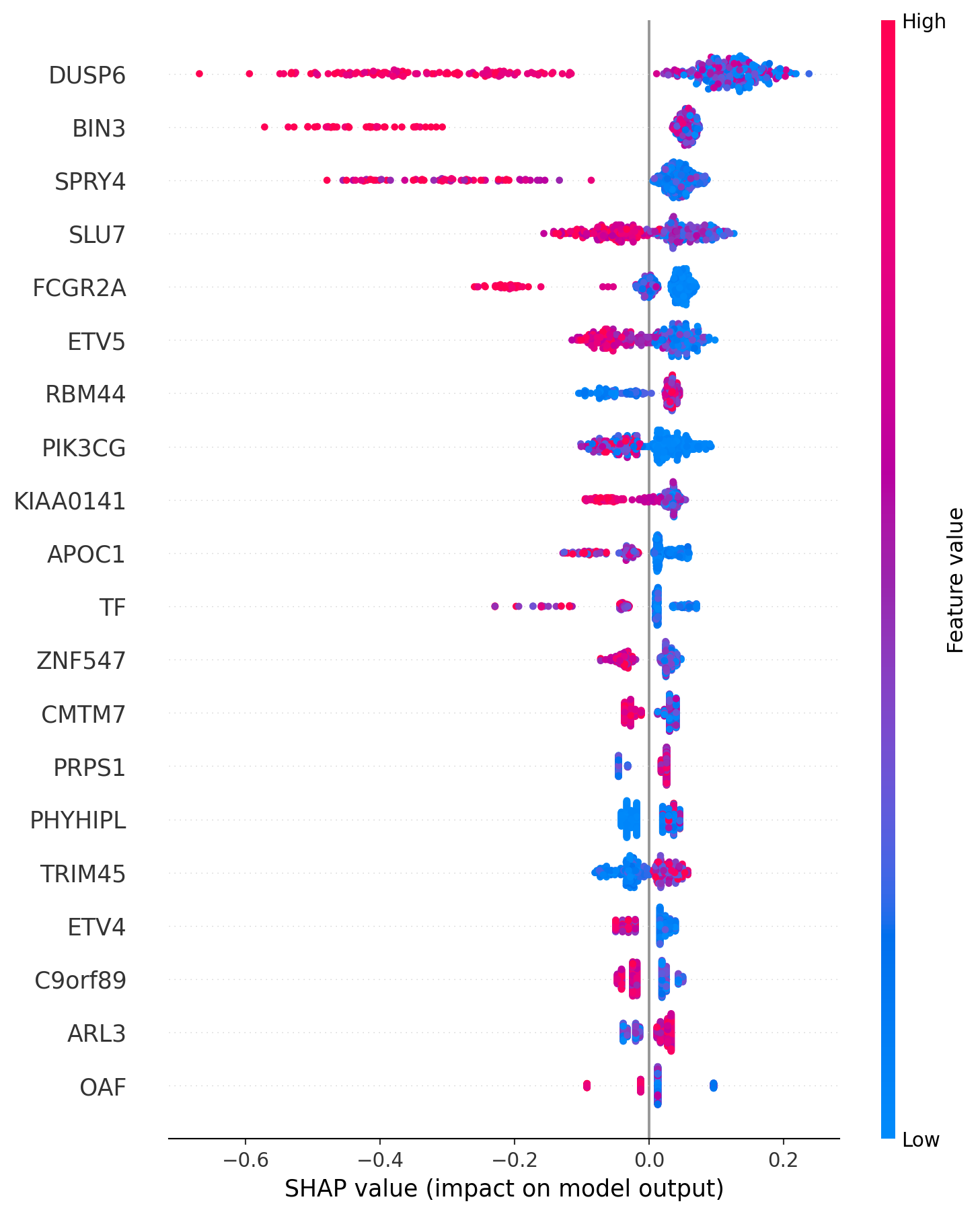
To get a little preview of what I’ll do later in4 this series, there’s a more powerful way of looking at how different variables affect prediction. This is using something called SHAP (SHapley Additive exPlanations) values. I’ll go more into these in another post, but here’s a plot of SHAP values for the model trying to predict AZ628 sensitivity.
Show the code
shap.initjs()
The colour of a point describes how high the feature (dependent variable) value is, and the x-position is the SHAP value. For example, a high DUSP6 expression value is associated with a more negative SHAP value, meaning a lower IC50, so the cell line is more sensitive to AZ628. There’s a lot more that can be done with these, but I think this is quite long enough for one post.
In subsequent posts, I’ll be aiming to improve model performance through various avenues, and trying to find whether we can actually make any useful interpretations of these models. They’re quite promising. For Refametinib, the model correlates really well with the observed data. This doesn’t take into account any factors other than changes in expression, so is quite impressive!
Footnotes
The International Transmembrane Society Symposium↩︎
by Emily Barnes at the MRC Laboratory of Medical Sciences↩︎
to be properly rigourous, we can’t really: correlation doesn’t imply causation! Without properly designed experiments this is all speculation↩︎